
Medical devices: the classification
An innovative sector, a secure market
Date
02/17/2023
An innovative sector, a secure market
Date
02/17/2023
At the crossroads of technology, medical devices are very much a part of our daily lives (compresses, crutches, monitoring applications, medical imaging equipment, glasses, syringes, etc.).
There are over 20,000 types of device (source: ANSM) that help to improve the quality of daily life, assist in the diagnosis of specific pathologies, or contribute to patient care and management. They are divided into several categories:
What exactly is a medical device?
Regulation (EU) 2017/745 (MDR)- Chap I, section 1 art. 2 gives us a detailed definition:
"Any instrument, apparatus, equipment, software, implant, reagent, material or other article, intended by the manufacturer to be used, alone or in combination, in humans for one or more of the following specific medical purposes:
and whose principal intended action in or on the human body is not obtained by pharmacological or immunological means or by metabolism, but whose function can be assisted by such means."
Not all medical devices are on the same footing: for example, from the use of a crutch to that of an artificial heart, the level of risk intensifies. These two medical devices will therefore not be subject to the same regulatory constraints in order to gain access to the market.
But what are the regulatory requirements?
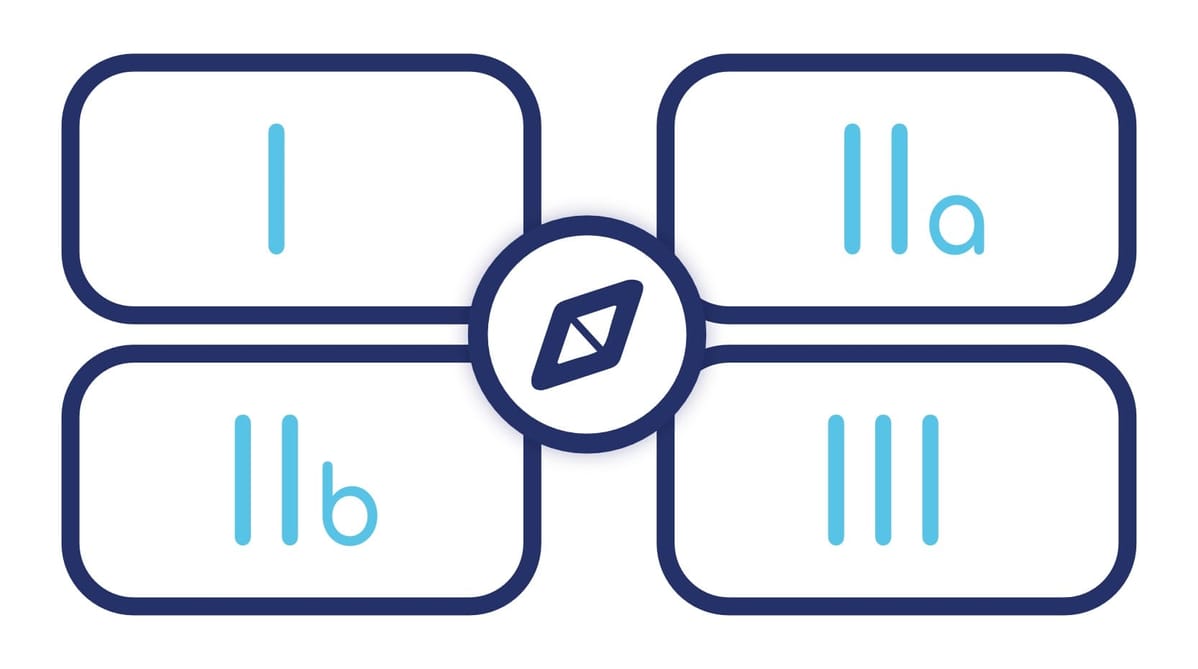
What are the different classes of DM?
DM are classified into 4 classes according to the level of risk associated with their use:
The higher the class, the more stringent the requirements.
Other types of medical devices exist alongside so-called "simple" medical devices: in vitro diagnostic medical devices. They have a specific classification and fall within a distinct regulatory reference framework ((EU) MDR 2017/746).
It is the manufacturer's responsibility to determine the class of his medical device according to the medical purpose for which it is to be used.
Identifying the class of a medical device is crucial to its further development and marketing. Initial misclassification could lead to longer lead times for compliance, or even prevent market access.
What criteria should you use to identify the class of your DM and meet regulatory requirements?
Classification rules are defined in Regulation (EU) 2017/745.
Medical devices are classified according to around 50 criteria, divided into 22 rules and 4 main families:
Please note: a medical device may belong to several families simultaneously. When in doubt as to the identification of its class, the highest risk class is preferred.
A medical device is classified according to several general factors:
• its expected useful life ;
• its application (invasive, implanted, requiring surgery) ;
• its type: whether it is an asset, i.e. a device requiring non-human energy (can also be therapeutic or diagnostic), software being an asset;
• If it operates on critical sites: central circulatory system or central nervous system;
and all aspects that may relate to the dangerousness of its use.
The impact of class on the CE marking procedure
The procedure for obtaining CE marking differs according to the class of medical device.
For Class I medical devices (lowest risk class), CE marking is obtained on the basis of a self-declaration issued by the manufacturer. In this case, the manufacturer must comply with the essential requirements for his product, and put in place the necessary means to ensure its performance and safety.
Within Class I devices, there are 3 Class I subcategories: (1) Class Im: medical device with a measuring function ; (2) Class Is: sterile
medical device ; (3) Class Ir: reusable surgical
medical device
These 3 sub-categories of medical devices are regulated differently from conventional Class I medical devices and require intervention of a notified body to obtain CE marking (audit of the specific part (measurement, sterilization, reuse))
For medical devices belonging to other classes (IIa, IIb, III), the procedure for obtaining CE marking is more restrictive and requires the implementation of 3 key elements:
In this case, CE certification is delivered by a notified body following one or more audits to assess product conformity.
The choice of organization is the responsibility of the manufacturer, who may seek assistance from a specialized body in compiling the necessary documentation.
Follow our newsletter, designed made for and by healthcare innovators !
In the program : our news, analyses, invitations, meetings in the DM and MedTech ecosystem...
This article is provided for information purposes only and does not constitute a normative or regulatory reference.





